
In case you haven't noticed, ChEMBL_18 has arrived. As usual, it brings new additions, improvements and enhancements both on the data/annotation, as well as on the interface. One of the new features is the target predictions for small molecule drugs. If you go to the compound report card for such a drug, say imatinib or cabozantinib, and scroll down towards the bottom of the page, you'll see two tables with predicted single-protein targets, corresponding to the two models that we used for the predictions.
- So what are these models and how were they generated?
They belong to the family of the so-called ligand-based target prediction methods. That means that the models are trained using ligand information only. Specifically, the model learns what substructural features (encoded as fingerprints) of ligands correlate with activity against a certain target and assign a score to each of these features. Given a new molecule with a new set of features, the model sums the individual feature scores for all the targets and comes up with a sorted list of likely targets with the highest scores. Ligand-based target prediction methods have been quite popular over the last years as they have been proved useful for target-deconvolution and mode-of-action prediction of phenotypic hits / orphan actives. See here for an example of such an approach and here for a comprehensive review.
- OK, and how where they generated?
As usual, it all started with a carefully selected subset of ChEMBL_18 data containing pairs of compounds and single-protein targets. We used two activity cut-offs, namely 1uM and a more relaxed 10uM, which correspond to two models trained on bioactivity data against 1028 and 1244 targets respectively. KNIME and pandas were used for the data pre-processing. Morgan fingerprints (radius=2) were calculated using RDKit and then used to train a multinomial Naive Bayesian multi-category scikit-learn model. These models then were used to predict targets for the small molecule drugs as mentioned above.
- Any validation?
Besides more trivial property predictions such as logP/logD, this is the first time ChEMBL hosts non experimental/measured data - so this is a big deal and we wanted to try and do this right. First of all, we did a 5-fold stratified cross-validation. But how do you assess a model with a many-to-many relationship between items (compounds) and categories (targets)? For each compound in each of the 5 20% test sets, we got the top 10 ranked predictions. We then checked whether these predictions agree with the known targets for that compound. Ideally, the known target should be correctly predicted at the 1st position of the ranked list, otherwise at the 2nd position, the 3rd and so on. By aggregating over all compounds of all test sets, you get this pie chart:

This is related to precision but what about recall of know targets? here's another chart:

This means that, on average, by considering the top 10 most likely target predictions (<1% of the target pool), the model can correctly predict around ~89% of a compound's known single protein targets.
Finally, we compared the new open source approach (right) to an established one generated with a commercial workflow environment software (left) using the same data and very similar descriptors:
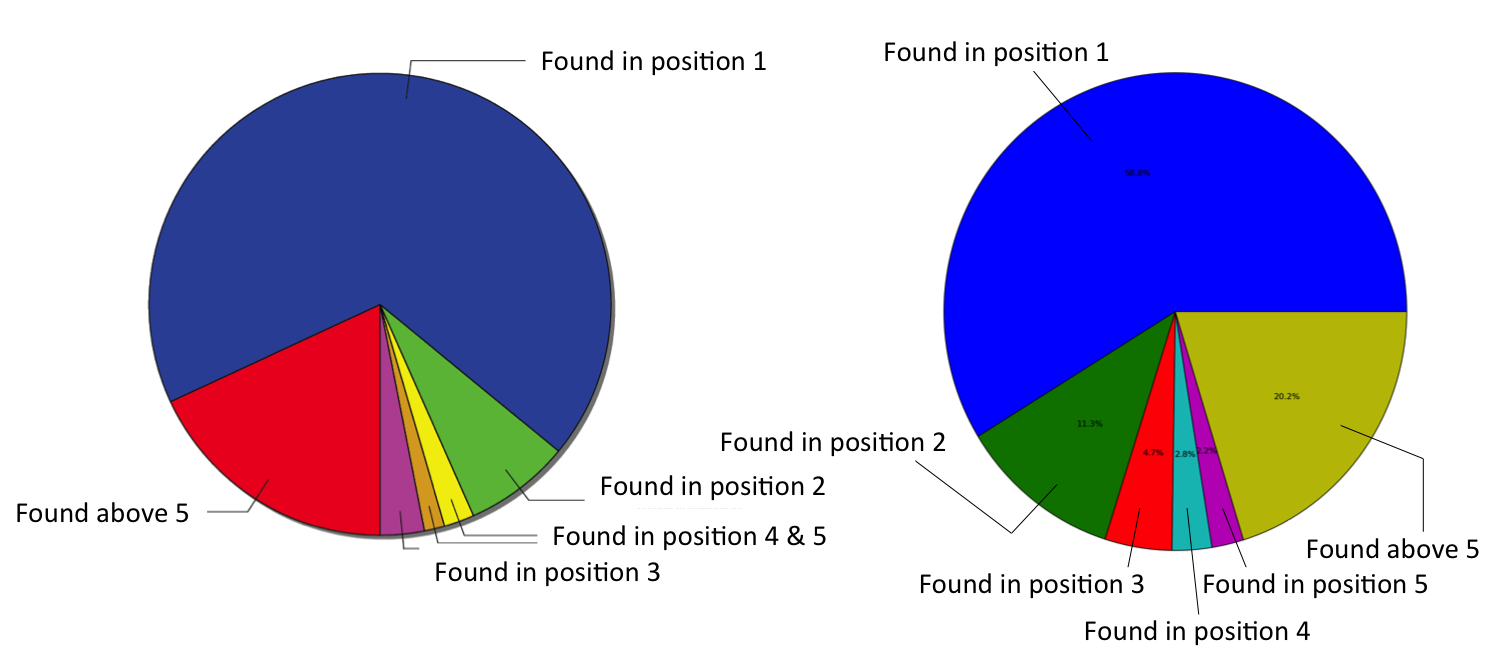
If you manage to ignore for a moment the slightly different colour coding, you'll see that their predictive performance is pretty much equivalent.
- It all sounds good, but can I get predictions for my own compounds?
We could provide the models and examples in IPython Notebook on how to use these on another blog post that will follow soon. There are also plans for a publicly available target prediction web service, something like SMILES to predicted targets. Actually, if you would be interested in this, or if you have any feedback or suggestions for the target prediction functionality, let us know.
George
Comments